
Apresentação:
- Marina Colella dos Santos
- Lara Garschagen
Comentários:
- Maria Almerinda V. F. Ribeiro Alves
- Pedro A. Gordan
- Edgard Torres dos Reis Neto
Às vezes, o diagnóstico clínico é um desafio. Em alguns casos, as peças representadas por sintomas, alterações em exame físico e propedêutica complementar não se encaixam nas primeiras tentativas de encontrar um diagnóstico e, dessa forma, estabelecer uma conduta adequada.
No meio médico, acredita-se que ao longo dos anos de carreira esse desafio torna-se mais fácil. Talvez pela comparação com um maior leque de manifestações clínicas e espectros de doença, por uma busca mais apurada dos detalhes ou pelo instinto clínico que se aguça.
A navalha de Ockham ou o princípio da economia é uma formulação científica na qual postula-se que dentre as explicações adequadas e possíveis para um determinado fenômeno seja escolhida a mais simples, aquela que contenha o menor número possível de variáveis e que sejam racionalmente lógicas para esclarecer o fato. Este é um dos pilares do reducionismo metodológico e que ganhou notoriedade no movimento minimalista, em vários momentos do século XX, muitas vezes sintetizado na expressão “less is more”, cunhada pelo arquiteto alemão Ludwig Mies van der Rohe, utilizada como título desta apresentação.
O caso clínico discutido foi um desafio e nos fez raciocinar e buscar conhecimento em todas as suas etapas. Segue um pouco do que foi discutido e levantado sobre o caso.
Caso Clínico
Mulher de 40 anos, branca, procurou nosso pronto-atendimento por quadro de aumento do volume abdominal e edema de membros inferiores com evolução para dispneia, ortopneia e quadros esporádicos de febre não aferida nos últimos 4 meses. Além disso, relatava diminuição do volume urinário no período, associado a episódios de urina avermelhada. Dois meses antes de nossa avaliação, ela foi internada em outro serviço para compensação do edema.
A paciente tinha como história patológica prévia uma doença de Wilson diagnosticada 13 anos antes que evoluiu com cirrose hepática Child B, esteatose e depósitos de cobre demonstrados em biópsia realizada após 1 ano do diagnóstico. Referia ter sido tratada com D-penicilamina por 6 anos e, posteriormente, com zinco por 3 anos, até 4 anos antes da internação, quando perdeu seguimento médico.
No momento da admissão, estava em uso de espironolactona 25 mg ao dia e furosemida 40 mg ao dia, além de fenobarbital 100 mg ao dia, por quadros de convulsões na infância, e vitamina C, que usava por conta própria.
A paciente era natural da Paraíba, mas morava em São Paulo havia 23 anos e trabalhava como auxiliar de limpeza de hospital. Filha de pais consanguíneos, referia história familiar importante, com seis irmãos falecidos pela mesma doença.
Era sedentária e negava tabagismo, etilismo e o uso de substâncias ilícitas.
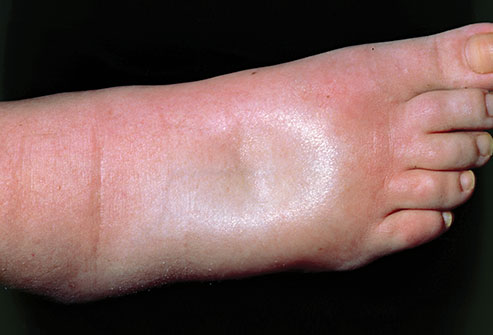
Sinais vitais e estado mental foram considerados normais na admissão hospitalar, enquanto ausculta pulmonar estava abolida em hemitórax direito e um edema moderado de membros inferiores até joelhos e ascite discreta estavam presentes.
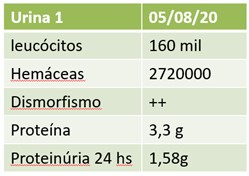
Na avaliação laboratorial, chamava atenção hipoalbuminemia de 2,6 g/L e proteinúria de 1,55g em urina de 24 horas, com volume urinário de 1380 mL. Em análise de sedimentos urinários, também se observava importante hematúria com 2.720.000 hemácias/mL, com dismorfismo eritrocitário, leucocitúria com 160.000 leucócitos/mL e proteinúria de 3,33 g/L. Função renal apresentava-se dentro da normalidade, com creatinina de 0,79 mg/dL e taxa de filtração glomerular estimada em 93,9 mL/min/1,73m² pelo método CKD-EPI.
A função hepática apresentava, além da hipoalbuminemia importante, transaminases pouco elevadas (TGO 55 e TGP 37) com coagulograma sem alterações. Hemograma com hemoglobina normal, linfopenia com contagem de leucócitos normais e uma plaquetopenia de 135 mil, como podemos avaliar nas Tabelas 1 e 2, abaixo.
TABELA 1
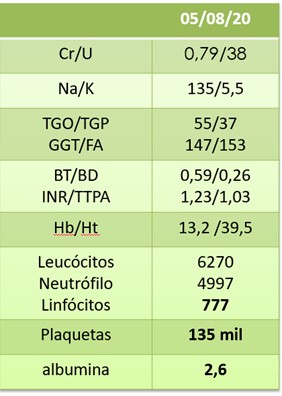
TABELA 2
Demais exames foram realizados durante a internação. A radiografia de tórax evidenciou extenso derrame pleural à direita, com derrame classificado como transudato após punção diagnóstica. Ecodopplercardiograma revelou função biventricular normal, sem evidência de derrame pericárdico. Ultrassonografia abdominal revelou sinais de hepatopatia crônica, pequena a moderada ascite e rins de tamanhos normais com relação corticomedular preservada.
Provas sorológicas para infecções virais foram solicitadas, com resultado negativo. Seu complemento sérico era consumido (C3 = 35, e C4 = 3). Apresentava como perfil auto-imune: fator antinuclear, anticorpos anti-DNA, anti-Sm, anti-SSa, anti-SSb, anti-RNP, anticardiolipina, anti-Jo1 todos negativos. Os valores de ANCA e crioglobulinas também eram negativos.
DISCUSSÃO DO CASO CLÍNICO
A paciente apresentava, portanto, um edema difuso de origem renal, já que não possuía descompensação hepática aguda para justificá-lo e tinha como achados positivos proteinúria subnefrótica, hematúria dismórfica, consumo de complemento e função renal preservada. Seus autoanticorpos eram todos negativos.
Dra. Lara: Com essa proteinúria subnefrótica associada a hematúria com função renal preservada, procuramos uma associação com a doença de base da paciente. Em uma revisão austríaca sobre a doença de Wilson1 encontramos uma correlação entre a d-penicilamina e proteinúria. Nela há relatos de casos com o aparecimento de glomerulopatia membranosa, lupus fármaco-induzido e, em altas doses, doença de anticorpo anti-membrana basal após o uso dessa medicação. 2 No entanto, a paciente estava sem medicação há 8 anos, sendo pouco provável. Como complemento é consumido, podemos ter uma glomerulonefrite membranoproliferativa (GNMP), associada a infecção ou a uma doença sistêmica.
Dr. Pedro: Inicialmente tive a impressão de que pudéssemos pensar em nefropatia por IgA secundária à cirrose, mas com esse complemento consumido, é pouco provável. Concordo com GNMP.
Dra. Almerinda: Deve-se descartar descompensação hepática relacionada a doença de base, o que não aparenta. Além disso, o que me chamou atenção foi o processo inflamatório capilar representado pela hematúria dismórfica e leucocitúria, provavelmente por uma glomerulonefrite proliferativa. Como é uma doença hipocomplementêmica, considero uma nefrite associada a infecção; não acredito em LES devido ao FAN negativo.
Dr. Edgard: O consumo de complemento pode ser secundário à própria cirrose da paciente por deficiência de produção dos fatores da cascata, o que permitiria o raciocínio de que todo o quadro renal poderia ser secundário a uma doença de IgA pela cirrose. Dessa forma, todos os achados estariam ligados a complicações da doença de base da paciente.
CONTINUAÇÃO DO CASO CLÍNICO:
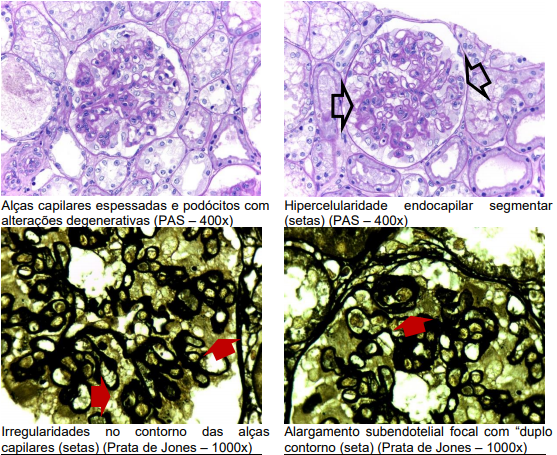
Com 6 dias de internação, indicou-se a realização de biópsia renal para elucidação diagnóstica, que revelou ao PAS 12 glomérulos, sendo 2 deles com discreta hipercelularidade endocapilar segmentar por células próprias e algumas células inflamatórias, alças capilares periféricas espessadas e com irregularidades no contorno subepitelial e imagens focais em duplo contorno, além de degeneração podocitária moderada; túbulos com focos de atrofia ao lado de fibrose intersticial discreta e ramos arteriais dentro da normalidade.
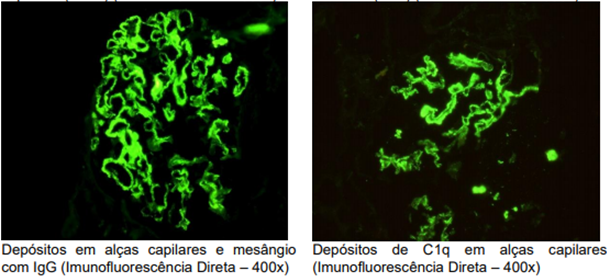
Em imunofluorescência direta, visualizados depósitos granulares de distribuição difusa, localizados em alças capilares e mesângio, com presença de predomínio de IgA (++) em associação com IgG (++), IgM (+), C1q (+/++), C3 (++) e cadeias leves Kappa (++) e Lambda (++).
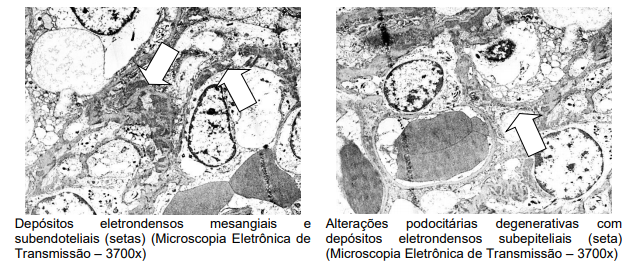
Já a microscopia eletrônica evidenciou depósitos eletrodensos mesangiais, subendoteliais e subepiteliais, além de hipercelularidade endocapilar discreta segmentar e alterações degenerativas podocitárias com retração de pedicelos.
Paciente apresentava, portanto, uma glomerulopatia membranosa com componente segmentar proliferativa com imunofluorescência full-house, mas com co-predomínio de IgA, sugestivo de doença sistêmica.
Microscopia óptica:
Imunofluorescência:
Microscopia eletrônica:
NOVOS COMENTÁRIOS SOBRE O CASO
Dra. Lara: Paciente com doença proliferativa e IF full-house. No entanto, FAN negativo e, pelos critérios EULAR/ACR publicados em 2019, FAN > 1/80 é uma condição de entrada. Temos codominância de IgA e poderia ser uma nefropatia por IgA secundária à cirrose, já há a redução do clearance dessa imunoglobulina, como já teorizado. Há ainda a possibilidade de associação com alguma infecção que não foi encontrada na investigação inicial.
Dra. Almerinda: A microscopia óptica me chama atenção pela pouca expressão de celularidade com predomínio de duplo-contorno, não podendo ser chamada de GNMP. A microscopia eletrônica descarta a associação de pós -infecciosa pelo padrão subepitelial dos depósitos.
Dr. Pedro: Em minha primeira análise do caso, tive como principal hipótese diagnóstica o LES. No entanto, revisando agora, acredito que todo o quadro clínico seja secundário à cirrose que a paciente já apresentava.
NOVAS INFORMAÇÕES DO CASO CLÍNICO:
Com o passar de 15 dias de internação, a paciente evoluiu com aparecimento de lesões papulares eritemato-violáceas em face, tronco, couro cabeludo e abdômen. Além disso, iniciou quadro de pancitopenia sem provas de hemólise.
Diante desse quadro sugestivo de doença sistêmica, foi solicitado anticorpo anti-P ribossomal, que apresentou positividade >200u/ml, o que nos permitiu o diagnóstico definitivo de Lúpus Eritematoso Sistêmico (LES).
Após 3 meses de seguimento, foi repetido o anti-DNA, que agora apresentou positividade de 1/20, como ocorre em alguns casos de LES FAN-negativo.
DISCUSSÃO ADICIONAL
Dra. Lara: Pelos critérios anteriores de LES, a paciente pontuaria. A classificação publicada em 1997 pelo American College Rheumatology (ACR) 4 era mais específica para a doença e apresentava 11 critérios, sendo que 4 eram suficientes para diagnosticar LES. Todos apresentavam o mesmo peso, e o FAN era um deles.
Em 2012, com o intuito de aumentar a sensibilidade e a precocidade do diagnóstico, a Systemic Lupus International Collaboration Clinics (SLICC) publicou uma nova classificação que ampliou os critérios, incluindo consumo de complemento e positividade de Coombs direto, e tornou obrigatório um critério imunológico. 5
Já a nova classificação EULAR/ACR de 2019, além de ter colocado o FAN positivo maior que 1/80 como critério de entrada, como já dito, requer a necessidade de 10 pontos com pelo menos um critério clínico. Há também diferentes pesos entre os sintomas.3
A paciente do caso preenche os dois critérios mais antigos para LES. No entanto, o critério mais novo não, já que não possui o FAN obrigatório.
Dr. Edgard: Os critérios foram feitos para aumentar a sensibilidade do diagnóstico e padronizar os pacientes para estudos científicos.
Há 1-2% de LES ativo com FAN negativo. Essa é a principal crítica ao critério EULAR/ACR, que, além de colocar o FAN como critério de entrada, também não diferencia o padrão do fator antinuclear. A caracterização do FAN é muito importante. Seus diferentes padrões sugerem a estrutura celular que está sendo acometida e há associações já conhecidas com determinados autoanticorpos ou mesmo com indivíduos saudáveis, o que leva ao risco do superdiagnóstico.
Por outro lado, o ponto que eu considero positivo da classificação de 2019 são os diferentes pesos entre os critérios.
Dra. Almerinda: LES é uma doença sindrômica, com diferentes origens fisiopatológicas. A doença de Wilson deposita cobre em célula epitelial, prioritariamente de epitélio tubular, mas também no podócito, alterando sua estrutura e podendo ser gatilho para formação de autoanticorpos que podem ser o início da fisiopatologia do LES dessa paciente.
Acredito também que o LES soronegativo mostra a doença autoimune sistêmica no início de sua imunogenicidade. Lembrando que essa paciente, por exemplo, apresentava linfopenia quando abriu o quadro de síndrome nefrótica.
Dr. Pedro: Na avaliação desse caso, a minha primeira hipótese era LES: síndrome nefrótica associada com nefrítica, consumo de complemento por via clássica, com função renal normal e IF full-house. O raciocínio clínico nos leva a pensar em um LES inicial. Em alguns casos devemos esperar a evolução da paciente para chegar ao diagnóstico definitivo. Em muitos casos, o FAN inicialmente negativo converte dentro de alguns meses.
Dra. Lara: O FAN é um conjunto de autoanticorpos descobertos na década de 40. Seu antígeno-alvo tem localização nuclear: em ácidos nucleicos, proteínas ou na cromatina. Ele pode ser realizado por 3 métodos: imunofluorescência indireta, multiplex ou ELISA. O mais sensível é a imunofluorescência indireta, que utiliza as células Hep2, em várias etapas do ciclo celular. 6,7
Em relação aos títulos, doenças autoimunes apresentam títulos moderados 1/160 ou elevados >1/640. Enquanto indivíduos saudáveis podem apresentar títulos baixos, até 1/80. Há associação entre infecções, medicações e neoplasias que também podem induzir autoimunidade transitória e gerar um FAN positivo em baixos títulos. 8,9
Existem também alguns autoanticorpos que não se ligam a moléculas no citoplasma, com anti-p ribossomal e, apesar de corarem na imunofluorescência direta, apresentam resultado de FAN negativo. Alguns estudos sugerem que mudássemos a nomenclatura para fator antinuclear.6
O Consenso Brasileiro de Fator antinuclear em células Hep-2 sugere a procura de outros anticorpos quando há suspeita clínica forte de LES e FAN negativo.
Dr. Edgard: Alguns anticorpos como anti-SSa, anti-SSb, anti-cardiolipina e o anti-P ribossomal correspondem a um FAN negativo. Existem ainda anticorpos que nós não conhecemos.
Quando anti-P ribossomal corresponde ao FAN positivo, é com mais frequência ao padrão pontilhado fino denso. Classicamente é associado a psicose e depressão, crises convulsivas, hepatite autoimune e glomerulopatia membranosa.
O anti-P ribossomal está presente em 10-30% dos pacientes com LES. Ele é pouco sensível mas muito específico, embora seja descrito em hepatite autoimune e doença de Chagas. Ele é usado como diagnóstico, mas não para seguimento clínico.
Dra. Lara: O anticorpo anti-P ribossomal tem possivelmente uma variabilidade, mas os estudos com caucasianos estimam uma prevalência entre 10-44% nos pacientes com LES e 70% de associação com anti-DNA. 10
Wang Y mostrou em um estudo observacional chinês que o anti-P ribossomal foi positivo em 27,9% dos pacientes estudados com diagnóstico de LES e que apresentavam negatividade para os outros autoanticorpos.
Há também relação do anti-P ribossomal com manifestações em idade mais jovens e lesões cutâneas, principalmente com eritema malar. No rim, apresenta função renal estável, com consumo e complemento e proteinúria.11
Do ponto de vista renal, há na literatura brasileira em que a positividade do anti-P ribossomal tem associação com nefrite lúpica classe V, cursando com mais proteinúria e função renal normal. Há alguns locais que sugerem que ele possa ser um possível marcador de componente membranoso. No entanto, 22% dos pacientes apresentavam também um componente proliferativo, como no caso da nossa paciente. 12
Dra. Almerinda: Ainda temos muito que trabalhar para fazer essa associação e usá-la como biomarcador. Mas é uma possibilidade. Seria uma das ferramentas para diagnóstico e tratamento individualizado.
Seria interessante até um estudo de anti-P ribossomal em outros casos de doença membranosa em pacientes não-lúpicas.
FINAL DO CASO E CONCLUSÕES:
Voltando ao caso, devido ao componente proliferativo renal e o comprometimento hematológico, optou-se por indução com corticosteróide e micofenolato de sódio.
No entanto, a paciente apresentou um quadro infeccioso pulmonar grave 4 meses após e acabou evoluiu a óbito.
O caso relatado nos fez entrar numa discussão muito rica durante sua condução, permitindo a elaboração de um raciocínio clínico com uma série de diagnósticos diferenciais.
No entanto, pudemos aprender também que às vezes menos é mais, e que perseguir o diagnóstico mais simples muitas vezes é o mais importante para o diagnóstico e o tratamento do doente.
REFERÊNCIAS BIBLIOGRÁFICAS:
- Roberts E, Schilsky A. A practice guideline on Wilson disease. Hepatology. 2003 Jun;37(6):1475-92.
- Ferenci P. Review: diagnosis and currenty terapy of Wilson disease. Aliment Pharmacol Ther. 2004;19:157–165.
- Aringer M et al. 2019 EULAR/ACR Classification Criteria for Systemic Lupus Erythematosus. Arthitis Rheum. 2019 sep;71(9): 1400-1412.
- Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997; 40:1725.
- Petri M et al. Derivation and Validation of Systemic Lupus International Collaborating Clinics Classification Criteria for Systemic Lupus Erythematosus. Arthritis Rheum. 2012 August; 64(8):2677–2686.
- Pisetsky D, Lipsky P et al. New insights into the role of antinuclear antibodies in systemic lupus erythematosus. Nature Reviews | Rheumatology. 2020.
- Dellavance A, Gabriel Junior A, Cintra AFU, Ximenes AC, Nuccitelli B, Taliberti BH et al. II Consenso Brasileiro de Fator Antinuclear em Células HEp-2. Rev Bras Reumatol. 2003;43(3):129-40.
- Olsen NJ, Karp DR. Autoantibodies and SLE—the threshold for disease. Nat. Rev. Rheumatol. advance online publication 3 December 2013.
- Dellavance A et al. Análise Crítica do Teste de Anticorpos Antinúcleo (FAN) na Prática Clínica. Rev Bras Reumatol. 2007;47(4):265-275.
- Choi MY et al. A review and meta-analysis of anti-ribosomal P autoantibodies in systemic lupus erythematosus. Autoimmunity Reviews. 2020;19(3):102463
- Wang Y, Luo P, Guo T, Zou L, Shi J, Chen P. Study on the correlation between anti-ribosomal P protein antibody and systemic lupus erythematosus. Medicine. 2020;99:20(e20192).
- Nascimento A. et al. Antibodies to Ribosomal P Proteins: A Potential Serologic Marker for Lupus Membranous Glomerulonephritis. Arthritis Rheum. 2006;54(5): 1568–72.

{kind=link}
{kind=link}